GM2 Gangliosidosis: Clinical Presentation
Lysosomal storage diseases have a broad diversity of clinical manifestations, but the most more commonly called Tay-Sachs disease, was one of the first identified lysosomal storage diseases. The severity of this disease varies among patients and is dependent on the degree of enzyme deficiency. The clinical phenotypes are generally grouped by the age of initial onset (infantile, late infantile, juvenile, and adult), but the disease can be more appropriately viewed as exhibiting a continuum of the “time of onset” from soon after birth to the second or third decade of life. In all forms, the dominant clinical presentation is neurological.
With the infantile form a retinal “cherry red spot” can be observed at birth. The child appears to develop normally for the first few months of life, but then both mental and physical abilities begin to rapidly deteriorate. The startle reflex becomes enhanced, the child becomes deaf and blind, and the condition continues to decline showing dementia and seizures. Death typically occurs by an age of five years. (Clarke 2002)
At the other end of the continuum, individuals with Adult Tay Sachs Disease, ATSD, also called Late Onset Tay Sachs (LOTS), typically develops mild ataxia and dysarthria by early teenage years. Physical coordination issues often become severe later in life. Vision and hearing remain intact, but patients may develop mental disorders including severe depression and bipolar disorder. Patients with the adult onset form typically have a normal life span. For more information, please refer to the related NIH Tay-Sachs web-site.
GM2 Ganglioside Storage
Tay Sachs disease is caused by the excessive storage of GM2 gangliosides in lysosomes. Gangliosides are a class of glycosphingolipid that consist of a ceramide backbone with three or more sugars esterfied to the ceramide alpha carbon and that also contain N-acetyl-neuraminic acid (sialic acid). The ganglioside name was selected based on the high concentration of these lipids found in ganglion cells, a type of neuron typically located near the inner surface of the retina of the eye. The GM2 ganglioside is composed of N-Acetyl-D-galactose-beta-1,4-[N-Acetylneuraminidate-alpha-2,3-]-Galactose-beta-1,4-glucose-alpha-ceramide. A perspective structure of GM2 ganglioside is shown in figure 3 and the corresponding ball-and-stick model is shown in figure 4. Shown in both of these figures is the stearoyl fatty acid form of ceramide.
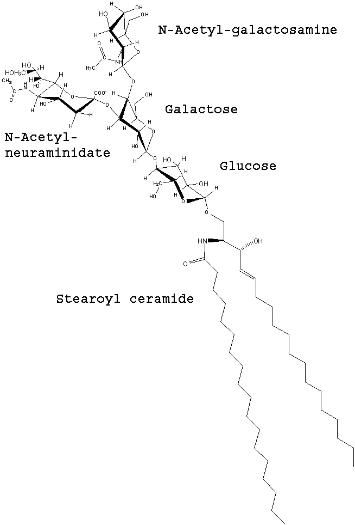

Figure 3. Structure of GM2 Ganglioside.
(Drawn by J. Keimel, ChemDraw software)
Figure 4. Model of GM2 Ganglioside.
(Drawn by J. Keimel, ChemDraw software)
Although several possible functions of GM2 ganglioside have been proposed, the role of these molecules has not yet been clearly established. The function may be an important consideration in the selecting a therapeutic option for GM2 gangliosidosis. As described later, one proposed therapeutic option is to reduce the synthesis of gangliosides with a small molecule inhibitor and thereby provide a balance between the synthesis rate and the available rate of catabolism. The clinical risks of excessive reduction in synthesis are related to the function of these molecules.
The structure and common location of these molecules has provided some clues regarding their function. The molecular shape suggests that the sialic acid and carbohydrate head of the molecule may extend from the plasma membrane while the fatty acid hydrophobic tails are anchored in the membrane. The exposed polar head of the molecule could thereby serve to interact with other membrane constituents or extracellular molecules. Based on an abnormal growth of neural cell dendrites in patients with high levels of GM2, as in Tay Sachs disease, it has been suggested that these molecules could be associated with dendrite initiation. There is also evidence that gangliosides along with other glycosphingolipids, cholesterol, and a variety of proteins appear co-located in specialized patches of cell membrane rather than being randomly distributed throughout the membrane. These specialized areas, called rafts, may represent transmembrane signaling platforms. Other studies suggest that these rafts may modulate proteins, such as growth factor receptors, associated with dentritogenesis (Walkley 2003).
The high concentration of these gangliosides in the central nervous system is likely the reason for the dominant neurological presentation of GM2 Gangliosidosis. While it has been observed that the GM2 synthesis rate fluctuates during brain development, the factors controlling this synthesis rate remains poorly understood. It may also be that the GM2 molecule is simply a degradation product of a more complex ganglioside on its catabolism pathway. The metabolic pathways for human sphingolipids with the detailed pathways of the ganglio-series are shown at the Kyoto Encyclopedia of Genes and Genomes web-site, https://www.genome.jp/pathway/map00604
Beta-Hexosaminidase
The lysosomal storage of acidic glycolipid GM2 gangliosides is the result of a deficiency in the enzyme activity associated with its degradation. The enzyme beta-N-acetylhexosaminidase A (Hex A, Enzyme Commission designation 3.2.1.52) and the associated small GM2 Activator protein are required for hydrolysis of the glycosidic bond and the removal of the terminal non-reducing N-acetylgalactosamine molecule from GM2 ganglioside to form GM3 ganglioside. See figure 3. The system has significant margin, and only large decreases in enzyme activity level cause clinical issues. A total activity level below 10% of normal is apparently required to elicit clinical symptoms. It has been observed that patients with enzyme activity levels near this value typically have the adult onset form of the disease, whereas patients with less than a few percent of normal values will manifest the infantile form. GM2 Gangliosidosis is inherited in an autosomal recessive manner. Individuals that are heterozygote carriers of a defective allele have less active enzyme than normal but more than the critical amount, and no symptoms have been attributed to this reduction in these individuals. A number of therapeutic options for GM2 Gangliosidosis are currently being explored which are intended to marginally increase enzyme activity levels above this critical activity level.
Hex A (112 KD) is a heterodimer with two subunits, designated as alpha and beta, which are approximately 60% similar to each other. Each subunit has only one known active site. (Lemieux 2006) Although the two subunits are similar, only the active site within the alpha subunit can adequately remove the N-acetylgalactosamine from GM2. Either of the two subunits can adequately hydrolyze beta-linked N-acetylgalactosamine and/or N-acetylglucosamine from a number of other molecules, but only the active site on the alpha subunit can remove the N-acetylgalactosamine group from GM2. The negatively charged sialic acid group (N-Acetylneuraminic acid) in GM2 prevents adequate interaction within the beta subunit. An appreciation for the possible interference being caused between the enzyme and the substrate can be gained by inspection of the substrate shape, figures 3 and 4. The protrusion of the sialic acid end-group apparently needs to be accommodated in the enzyme structure in order for the N-Acetyl-glucosaminidase to enter the active site.
The GM2 Activator protein assists in the transport of the GM2 molecule from the membrane and transports the GM2 to the lysosome. Defects in the activator protein are also known to cause GM2 lysosomal storage possibly from the inability for the GM2-GM2 Activator protein complex to adequately dock with the Hex A heterodimer leaving the substrate to accumulate in the lysosome.
The alpha subunit has a mature sequence of 492 amino acids and the beta subunit has a mature sequence of 489 amino acids. Each subunit consists of two domains, Domain I and II. The function of Domain I is unknown. Domain II contains the active site for each subunit and has a primary tertiary structure consisting of a TIM barrel fold (protein fold consisting of eight α-helices and eight parallel β-strands). The active sites are located at the opening of the TIM barrel at the interface between the alpha and beta subunits. As depicted in Figure 5, the αGlu323 (glutamate amino acid number 323 on the alpha subunit) is the general acid-base residue for protonation of the glycosidic bond. The αAsp322 provides a negatively charged carboxylate group that stabilizes the positive charge of the C1 nitrogen atom of the oxazolinium ion intermediate (Lemieux 2006).
While the active site is focused in just one domain of the alpha subunit, defects in either of the Hex A subunits or in the GM2 Activator protein are known to cause GM2 gangliosidosis. There are more than 92 different genetic mutations known to cause the disease. (Scriver 2001)
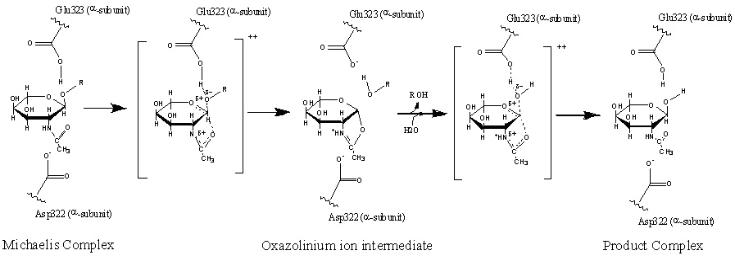
Figure 5. The proposed catalytic mechanism for GM2 ganglioside being converted to GM3 ganglioside is shown. (Adapted from Mark 2003. Re-drawn by J. Keimel using ChemDraw software.)